Research
The extraordinary re-patterning of gene expression that occurs during cellular differentiation dictates the identity of a resultant cell-type. This gene expression profile is then inherited by daughter cells upon cell division, ensuring that this cellular identity is sustained. Whether genes are repressed or active in transcription boils down to the structure of chromatin; its accessibility or not to the transcriptional machinery. Features that foster the chromatin structure without affecting the DNA sequence and that are inherited (see below) during cell division are “epi”genetic. Some of the post-translational modifications (PTMs, e.g. methylation, ubiquitination, amongst others) of the histone components of the nucleosomes may be epigenetic as the factors that catalyze and/or recognize these modified histones foster appropriate chromatin structures. For example, the Polycomb group (PcG) of proteins maintain gene repression by impacting the chromatin state. PcG proteins exist in two major multi-subunit Polycomb Repressive Complexes (PRC) in the cell: PRC1 and PRC2 (see below). The different chromatin states are further organized into a three-dimensional architecture. CTCF is a major protein constituent of this architecture as it insulates repressed from active chromatin domains (see below).
Our studies explore the formation of chromatin structure and the changes to chromatin architecture. We use a multi-disciplinary approach to understand how the proteins and protein complexes described below, contribute to the normal differentiation process and how naturally occurring mutant forms of some of these proteins thwart a normal cellular identity. Our approaches include state-of-the-art technologies (e.g. ChIP-seq or related sequencing methods, RNA-seq, single-cell analyses, microscopy, proteomics, mouse genetics and phenotypic analyses, gastruloids for developmental analyses, and any technology necessary to advance our research). Oftentimes, we seek cellular conditions to capture the initiation of the process understudy, rather than its features at equilibrium in “steady state” cells. By tracking these processes de novo, we have learned about key events that otherwise would have been obscured. As evident in several examples below, when repression is aberrantly lost from a chromatin domain, that domain becomes engaged in transcription corrupting the normal gene expression profile and associated cellular identity.
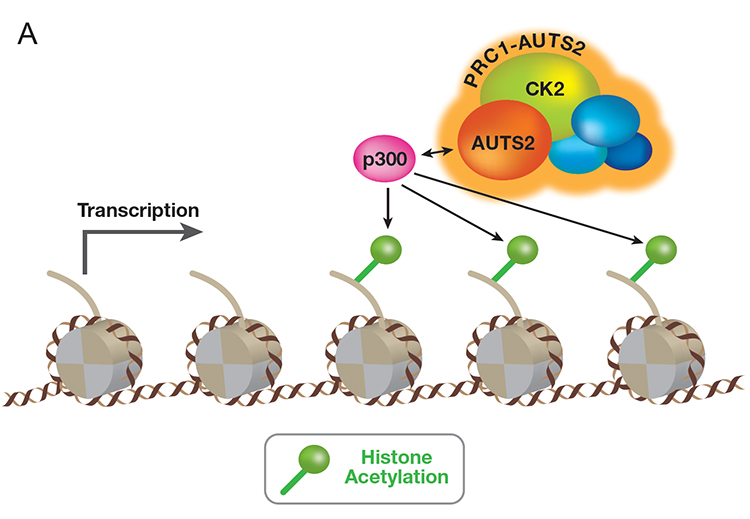
Mammalian PRC1 complex comprises a subset of Polycomb group (PcG) proteins that maintain transcriptional repression programs. Our proteomic analyses revealed that PRC1 complexes actually form minimally six major families. Characterizing the distinct composition of each family revealed that two sub-families (PRC1-AUTS2) contain three novel, non-PcG constituents: 1) AUTS2, 2) the transcription co-activator P300, and 3) the protein kinase CK2. AUTS2 is often disrupted in patients with neuronal disorders, yet the mechanism underlying such pathogenesis is unclear. Contrary to the well-established role of PRC1 in repression, PRC1-AUTS2 unexpectedly activates gene expression. The mechanistic basis of this phenomenon involves CK2-mediated inhibition of PRC1 repression and AUTS2-mediated recruitment of the transcription coactivator, P300. The recruited P300 facilitates transcription in part by acetylating the histones (Figure A, below).

& Inroads into Disease
Our findings pointed to the role of AUTS2 in positively regulating the central nervous system (CNS) transcriptional program: conditional targeting of AUTS2 in the mouse CNS led to various developmental defects. Our latest findings demonstrate that patients harboring mutations in the HX repeat domain of AUTS2 protein exhibit defects in AUTS2 interaction with P300, as well as a developmental disorder reflective of Rubinstein-Taybi syndrome. The absence of AUTS2 or mutation in its HX repeat domain gives rise to a mis-regulation of a subset of developmental genes and curtails motor neuron differentiation of mouse embryonic stem cells. Moreover, the transcription factor, Nuclear Respiratory Factor 1 (NRF1) exhibits a novel and integral role in the normal neurodevelopmental process by recruiting PRC1-AUTS2 to chromatin (Figure B, above).
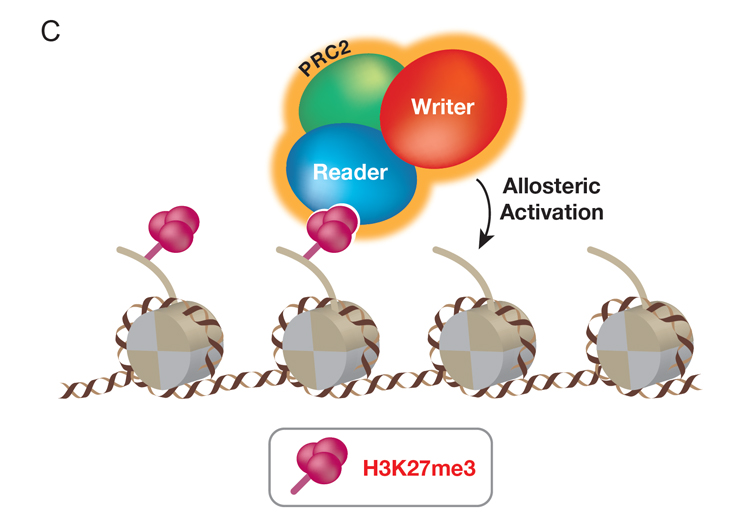
Mammalian PRC2 complex comprises a subset of PcG proteins different from those of PRC1. PRC2 catalyzes tri-methylation of lysine 27 of histone H3 (H3K27me3), which is associated with repressed chromatin. Previously, we discovered that PRC2 binds to its own catalytic product, H3K27me3, resulting in its allosteric activation and promoting its further catalysis of H3K27me3. This “write and read” mechanism is instrumental to the generation of extensive, repressed regions of chromatin containing H3K27me3 in the cells. Key to PRC2 function is its recruitment to the proper chromatin sites. We designed an in vivo system to capture PRC2 in action as it establishes de novo its repressive chromatin domains. A subunit of PRC2 was genetically deleted such that endogenous PRC2 and H3K27me3 levels were depleted in these cells. This subunit is then inducibly re-expressed such that the initial presence of PRC2 and its activity on chromatin can be tracked. The initial and primary PRC2 recruitment sites correspond to the promoters of developmental genes and through knockout experiments, we identified which of its partner proteins are required for PRC2 recruitment. By tracking PRC2 and its catalysis of H3K27me3 in chromatin in this manner, the requirement for the PRC2 “write and read” mechanism for spreading H3K27me3 is evident. This system can be adapted to differentiating cells wherein PRC2 is critical for lineage integrity and its recruitment might entail other important partner proteins yet to be identified (Figure C, below).
& Inroads into Disease
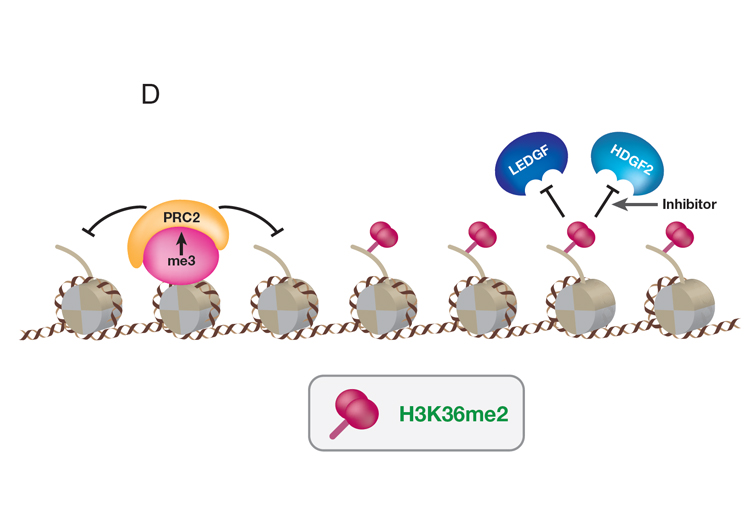
Remarkably, Diffuse Intrinsic Pontine Gliomas (DIPG), an untreatable pediatric cancer, contain a lysine to methionine mutation (H3K27M) in the PRC2 substrate (H3K27). While H3K27M is present in only one of 15 genes encoding histone H3, its considerable impact on PRC2 is evidenced by the widespread loss in the product of PRC2 catalysis (H3K27me3). To understand this failure in PRC2 function, we induced expression of H3K27M in tissue culture cells and followed its incorporation into chromatin and its effect on PRC2. While PRC2-H3K27M interaction is transient, PRC2 is released in a persistent, functionally less active state, being compromised in its ability to spread H3K27me3 appropriately throughout the genome. This loss in H3K27me3 at its normal chromatin sites is met with an inappropriate invasion of histone post-translational modifications associated with active genes, overturning the normal gene expression profile. This invasion includes H3K36me2. We discovered that two proteins that bind to H3K36me2, LEDGF and HDGF2, facilitate the transcription process. These discoveries have implications for therapeutic intervention by targeting LEDGF and HDGF2 in DIPG (Figure D, above).
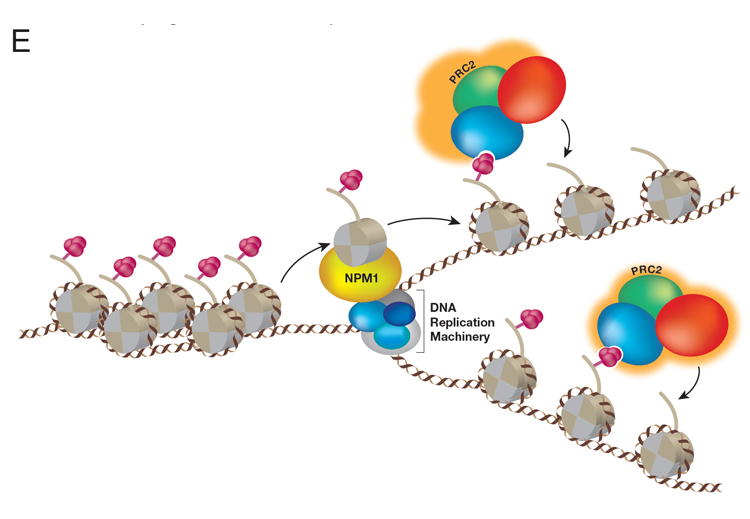
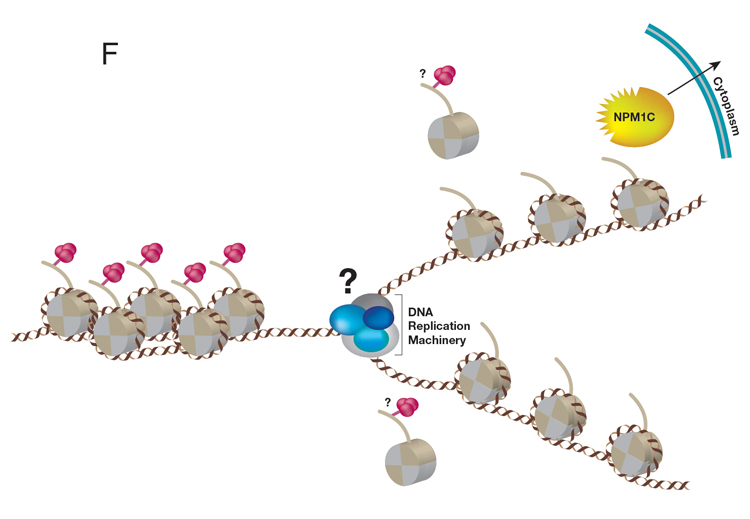
A central question in the field of Epigenetics/Mammalian Gene Regulation is how a particular cellular identity is properly inherited by progeny cells during cell division. But first, which type of chromatin domain is inherited during DNA replication: chromatin active in transcription or transcriptionally repressed or both? To address this fundamental process that preserves cellular identity, we designed a unique and powerful new approach that captures parental nucleosome inheritance as it occurs in vivo, at single gene resolution during DNA replication. This system uses endogenous histone H3 that is primed for biotin-tagging. Upon targeting a novel version of dCas9 exclusively to a single locus of choice using gRNAs, the histones are biotin-tagged within proximal nucleosomes before DNA replication. The segregation of these nucleosomes from this selected single locus one that is either active or repressed for transcription is then tracked during DNA replication. This system revealed the crux of epigenetic inheritance: nucleosomes are re-deposited locally, to the same chromatin domains, in the case of replicating DNA from repressed, but not active genes; nucleosomes from active chromatin domains scatter. Thus, only features inherent to repressed chromatin are inherited. We further discovered that a previously characterized histone H3/H4 chaperone, NPM1, actually associates with replicating repressed chromatin and is required for its inheritance (Figure E, below).
& Inroads into Disease
Of great importance to our future studies of NPM1 is its oncogenic potential. NPM1 mutations altering human NPM1 protein at its C-terminus (NPM1c), displace NPM1 from its normal locales, the nucleolus and nucleoplasm, to the cytoplasm. NPM1c, is found in ~35% of all Acute Myeloid Leukemia (AML) with normal karyotype. Notably, NPM1c AML cells exhibit abnormal de-repression of the developmentally important HOX genes, resulting in HOX proteins driving leukemogenesis. Importantly, the best-studied and developmentally-relevant target genes of PRC2 are the HOX clusters. The loss of gene repression and dysregulation of epigenetic mechanisms are hallmarks of cancer. We speculate that an NPM1-PRC2 axis may be misregulated during the development of AML and are investigating the functional interplay between PRC2 and NPM1c during AML leukemogenesis (Figure F, above).
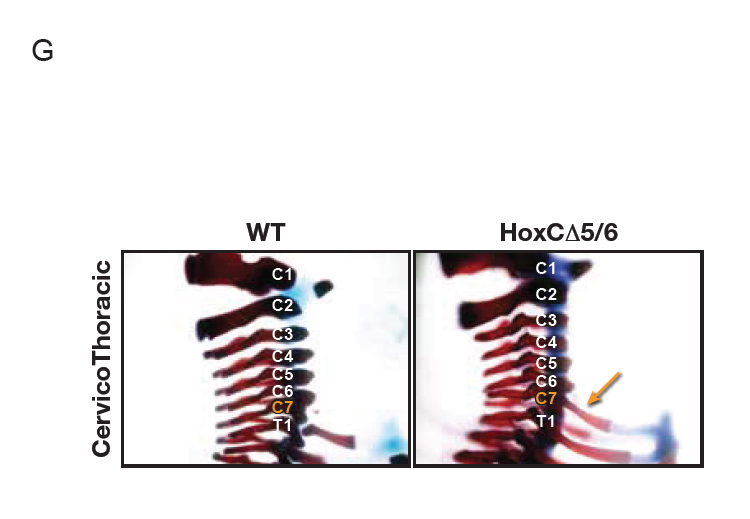
We sought to understand how PRC2 activity is closed off from actively transcribed genes and we found that CTCF is critical to this process: a single CTCF chromatin-binding event spatially organizes the HoxA cluster to shield PRC2 from neighboring regions of active gene expression during differentiation of mouse embryonic stem cells into cervical motor neurons. In accordance with the HOX clusters being paramount to development and the key role of CTCF in this process, deletion of this CTCF binding event disrupts the chromatin boundary such that active transcription invades the normally repressed domain, leading to homeotic transformations in the mouse (Figure G, below). Thus, CTCF functions to insulate repressed from active chromatin domains by establishing appropriate topological boundaries; of critical importance to proper differentiation.
& Inroads into Development
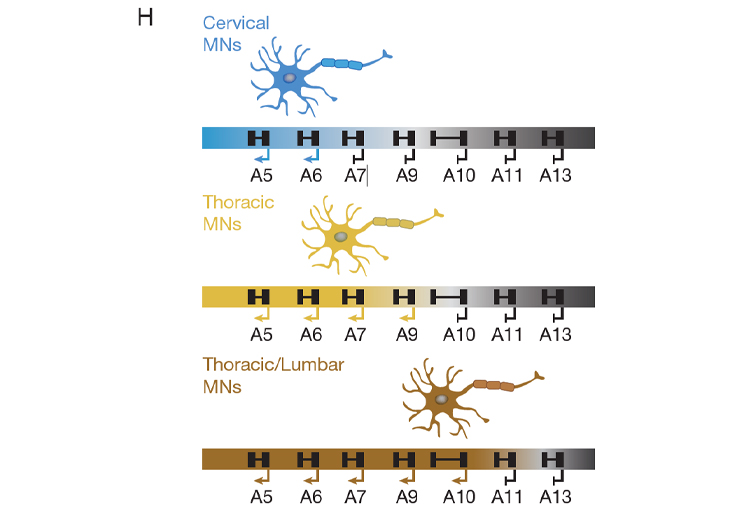
As we continue to investigate exactly how CTCF performs this critical boundary function, we can surmise that CTCF does not act alone, as CTCF at a particular DNA-binding site functions as an insulator in one cell type, but not in another. Indeed, through several distinct approaches, we identified and verified the existence of novel insulators that function similarly to CTCF, but at distinct DNA sites within the HOX clusters. Remarkably, these insulators are differentially expressed across different tissues and cell-types. This scenario points to the existence of a network of insulating factors, other than CTCF, that function in a combinatorial manner along with CTCF to organize appropriate chromatin architecture and thus, foster appropriate gene expression patterns during the processes of differentiation. Developing a solid understanding of these variations in chromatin activity at the molecular level will be instrumental to fully understanding the gist of the developmental program (Figure H, above).